rDock: a fast, versatile, and open-source program for docking ligands to proteins and nucleic acids¶
rDock is a fast and versatile open-source docking program that can be used to dock small molecules against proteins and nucleic acids. It is designed for high-throughput virtual screening (HTVS) campaigns and binding mode prediction studies.
rDock is mainly written in C++ and accessory scripts and programs are written in C++, Perl or Python languages.
The full rDock software package requires less than 50 MB of hard disk space and it is compilable (at this moment, only) in all Linux computers.
Thanks to its design and implementation [rDock2014], it can be installed on a computation cluster and deployed on an unlimited number of CPUs, allowing HTVS campaigns to be carried out in a matter of days.
Besides its main Docking program, the rDock software package also provides a set of tools and scripts to facilitate preparation of the input files and post-processing and analysis of results.
About¶
Download¶
Please visit rDock GitHub page for most up to date releases.
Features¶
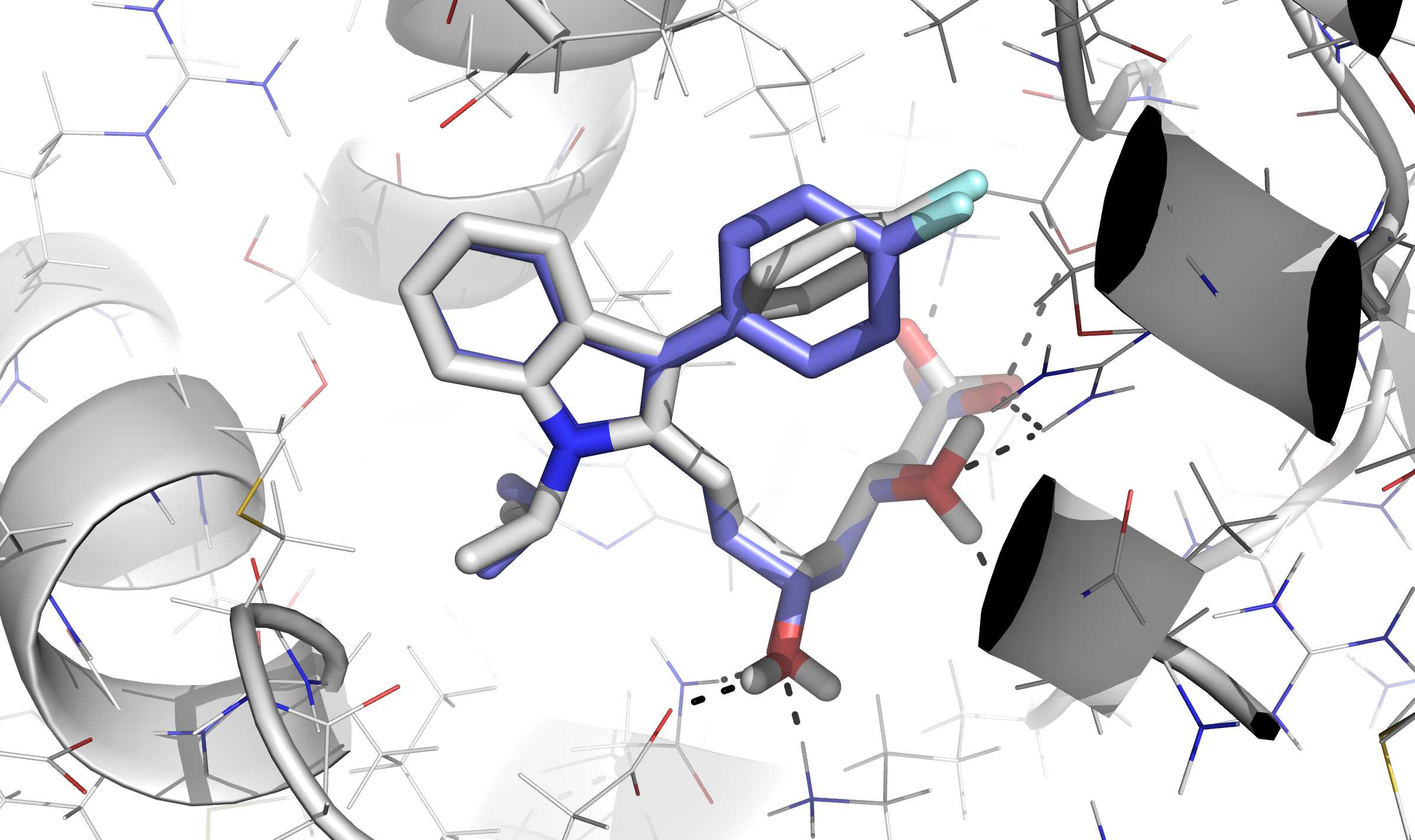
Fig. 1 The above image illustrates the first binding mode solution for ASTEX system 1hwi, with an RMSD of 0.88 Å.¶
- Docking preparation
Define cavities using known binders or with user-supplied 3D coordinates. Allow -OH and -NH2 receptor side chains to rotate. Add explicit solvent molecules and structural waters. Supply pharmacophoric restraints as a bias to guide docking.
- Pre-processing of input files
Define common ligand structure for performing tethered docking (requires Open Babel Python bindings). Sort, filter or split ligand files for facilitating parallelization. Find HTVS protocol for optimizing calculation time. Pre-calculate grids to decrease subsequent calculation times.
- Post-processing and analysis of results
Summarize results in a tabular format. Sort, filter, merge or split results files. Calculate RMSD with a reference structure taking into account internal symmetries (requires Open Babel Python bindings).
- Binding mode prediction
Predict how a ligand will bind to a given molecule. The ASTEX non-redundant test set for proteins and DOCK and rDock test sets for RNA have been used for validating and comparing rDock with other programs.
- High-throughput virtual screening
Run for million of compounds in short time by exploiting the capabilities of computer calculation farms. Ease of parallelization in relatively unlimited CPUs to optimize HTVS running times. The DUD set has been used for validating rDock and comparing its performance to other reference docking programs.
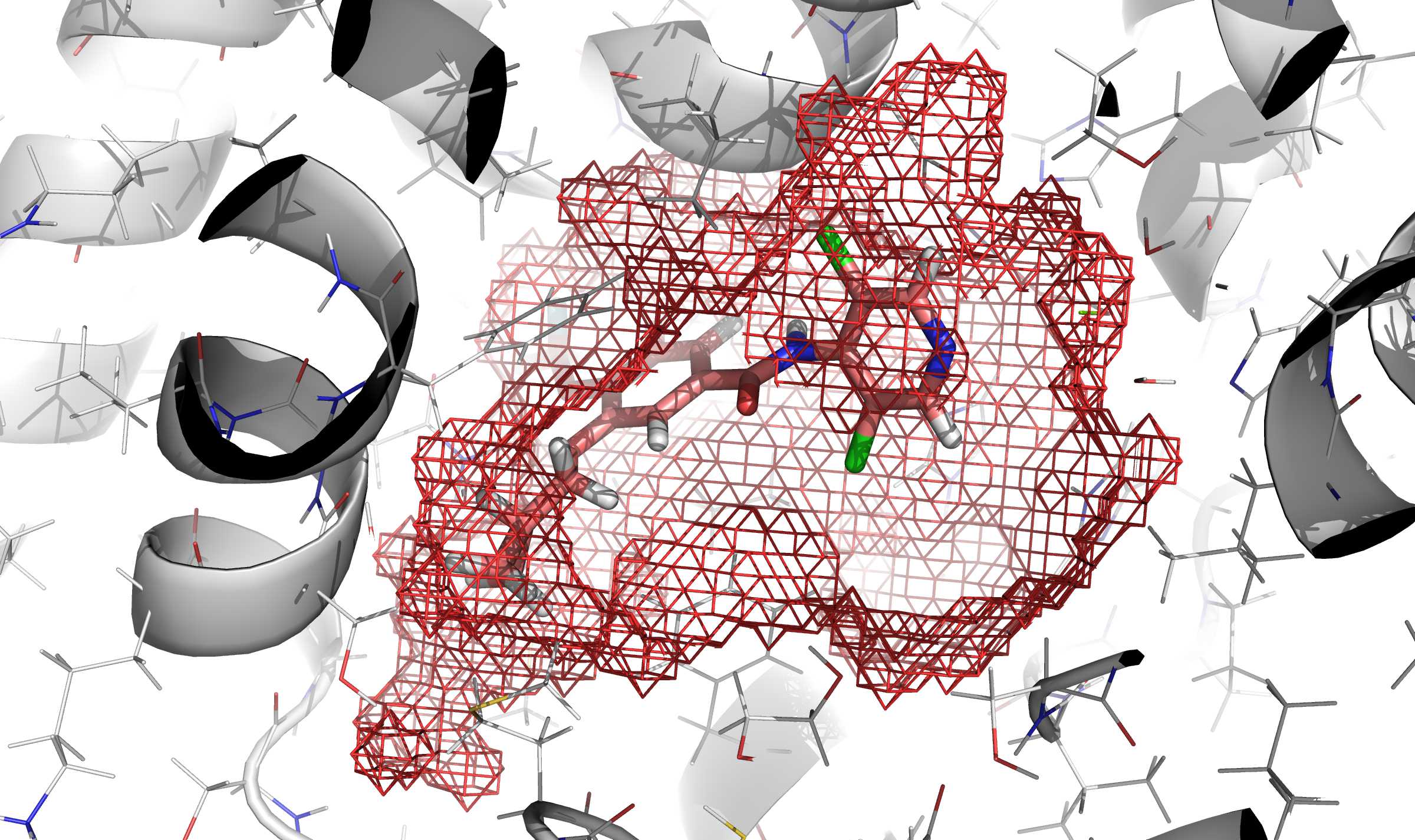
Fig. 2 In red mesh, definition of the cavity obtained by execution of rbcavity
program.¶
History¶
The RiboDock program was developed from 1998 to 2006 by the software team at RiboTargets (subsequently Vernalis (R&D) Ltd) [RiboDock2004]. In 2006, the software was licensed to the University of York for maintenance and distribution under the name rDock.
In 2012, Vernalis and the University of York agreed to release the program as open-source software [rDock2014]. This version is developed with support from the University of Barcelona – github.com/cbdd/rdock.
License¶
rDock is licensed under GNU LGPL version 3.0.
References¶
If you are using rDock in your research, please cite [rDock2014]. Former software reference provided for completeness is [RiboDock2004].
Ruiz-Carmona, S., Alvarez-Garcia, D., Foloppe, N., Garmendia-Doval, A. B., Juhos S., et al. (2014) rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput Biol 10(4): e1003571. doi:10.1371/journal.pcbi.1003571
Morley, S. D. and Afshar, M. (2004) Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock®. J Comput Aided Mol Des, 18: 189–208. doi:10.1023/B:JCAM.0000035199.48747.1e
Getting started guide¶
In this section, you have the documentation with installation and validation instructions for first-time users.
To continue with a short validation experiment (contained in the Getting started guide), please visit the Validation experiments section.
Reference guide¶
In this section you can find the documentation containing full explanation of all rDock software package and features.
For installation details and first-users instructions, please visit Installation and Getting started sections.
Tutorials¶
Support¶
If you are having some trouble regarding usage, compilation, development or anything else, you can use different options to ask for support.
Mailing lists¶
If you are having any kind of trouble, you have any questions or anything related to general usage of the program please search and use our mailing lists.
Issue tracker¶
Please use GitHub Issues for any comments or assistance needed.
